![]()
![]()
![]()
![]()
Prof. Dr. Rossitza Pentcheva
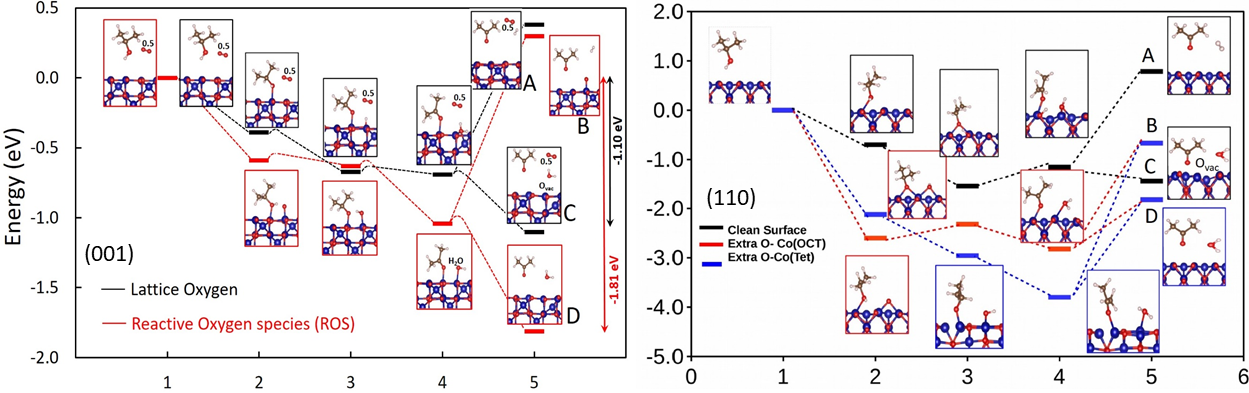
A key aspect of research within this project is to provide theoretical understanding and identify trends in the catalytic activity within a series of transition metal oxides as a function of structural motifs, crystallographic orientation, and doping level. Based on large-scale density functional theory calculations (DFT) taking into account electronic correlations beyond the local density approximation, we explore selected oxidation processes (e.g., OER, isopropanol) on spinel and perovskite surfaces with different crystallographic orientation, termination and reaction sites. The goal is to establish a relation between the energetics of adsorbates and intermediates and the underlying structural and electronic properties and to identify potential active sites. A main incentive for the second funding period is the extension towards considering multisite mechanisms, kinetic effects, and the effect solvation (both implicit and explicit). Moreover, we will address the role of hydroxide formation on the OER activity. The project aims to provide fundamental understanding in the underlying mechanisms in the two materials classes synthesized and characterized within the experimental projects (C01-C03, B02, B03, B09), especially the ones working with thin oxide films (projects A04, A07), as well as the ones investigating the catalytic properties of these surfaces (A01, A02, A09).
(Figure: Energetics of intermediates during 2-propanol to acetone oxidation with respect to the initial configuration (2-propanol in the gas phase) at the (left) (001) and (right) (110) Co3O4 surface. Four mechanisms were studied: in A and B acetone desorbs from the surface together with H2, and in C and D the products are acetone and water. In C an oxygen vacancy is created at the surface (Mars–van Krevelen mechanism), and in D the oxygen of the desorbed water stems from coadsorbed oxygen on the surface. In the intermediate stages, hydrogen from the deprotonation of 2-propanol binds at a lattice oxygen or OH group (A, C) or at an adsorbed oxygen (D). In order to compare the relative stability of intermediates for the four pathways, 0.5EO2 is added to the total energy for paths A and C).

Copyright © TRR247 2023
Last update: Jun 27, 2023