![]()
![]()
![]()
![]()
Prof. Dr. Frank Neese
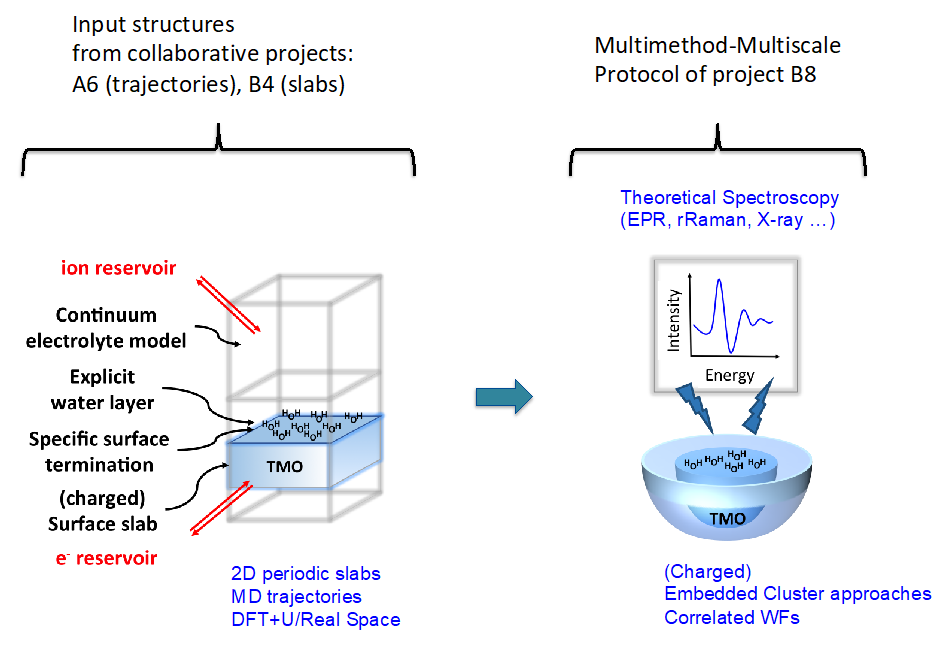
The determination of an accurate atomistic structure of a catalyst under operando conditions that involves charged surfaces in contact with an electrolyte is a highly difficult and challenging problem implying that a combined multimethod-multiscale theoretical approach is urgently needed. However, theoretical methods for such conditions are not readily available and extensive method development is required. This was fully explored during the first funding period (FP1). In the initially formed tandem project of two groups (Neese and Schmid) with complementary expertise, a multimethod approach was developed that involves accurate computational prediction of spectroscopic data of the actual solvated catalytic active sites, by combining periodic slab and embedded cluster computational approaches. This implies on one hand close interaction with the complementary other theoretical projects. On the other hand, close collaboration with experimental projects investigating spectroscopic response is pivotal.
For the second funding period (FP2) the project is based strongly on the methods and the computational workflow, which was developed in the FP1 but also on the experimental information on the catalyst systems that has been gained in FP1. In FP1, the Neese group developed a series of wavefunction based protocols that are adequate in treating the high complexity of the Co3O4 oxide surface models. This involves 1) the development of a general spin Restricted Open Shell Configuration interaction approach ROCIS that is able to compute various X-ray spectra of both high spin (HS) and antiferromagnetically coupled (AF) systems with high predictive accuracy. 2) A complete redesign of the embedded cluster approach protocol to now operate under a QMMM framework. 3) The development of a wavefunction based protocol that is able to predict the X-ray emission spectra of the Co3O4 oxide surface models in collaboration with the DeBeer group in the framework of the B06 project. Within the FP1 a combined wavefunction based and DFT based protocol that can investigate the Band Gap problem of the Co3O4 oxide surface models was developed. This is based on the complete active space iterative configuration expansion (CAS-ICE-CI) wavefunction method that was developed in the Neese group, and it is able evaluate the performance of the DFT protocols to cross correlate the results with the other theory groups. We anticipate that these developments during the FP1 will strongly aid the catalyst structure investigations during the FP2.
Studying the spectroscopic response of reactive surfaces in the framework of the embedded cluster approach requires the availability of sufficient large clusters that can correctly describe the short and long-range effects of the real surfaces. While in our family of methods clusters with about 200-300 atoms (including 10-20 metal centers) can be routinely treated, in the timeframe of FP2 we plan to implement a linearly scaling version of the QM/MM interaction potential terms in the concept of the fast multipole methods (FMM). This will allow even larger cluster sizes to be treated using wavefunction based methods, that are adequate to study the surface catalytic activity under reaction conditions and probe the spectroscopic response of important reaction intermediates with high predictive ability.
One of the major experimental evidence of the FP1 is the formation of an amorphous Co(III)-hydroxyde layer forming before and under OER conditions form Co3O4 surfaces. This means that “ideal” Co3O4 surfaces generated by simply slicing bulk is not a valid catalyst model. One fundamental target is the theoretical investigation of the transformation (i.e., oxidation) of the cobalt oxide into the hydroxide under a positive bias potential. There are in principle three fundamental processes during such catalytic reactions: 1) the radical species formation 2) surface oxidation and 3) the catalytic species spin and oxidation states. Together with the Spohr group (A06) we plan to study such reactions in the time domain using a combined protocol that involves solvated slabs, periodic molecular dynamics simulations and embedded cluster calculations. Optical (X-ray) spectroscopic fingerprints and magnetostructural correlations will be computed from the trajectories for relevant intermediates in order to explore these important processes in collaboration with the corresponding experimental groups. This also involves a systematic library of structural-spectroscopic correlation that needs to be developed and employed to identify the active species of the working catalyst.
(Figure: Schematic representation of the Multimethod Multiscale protocol of project B08).

Copyright © TRR247 2023
Last update: Jun 27, 2023