![]()
![]()
![]()
![]()
Prof. Dr. Kai S. Exner • Prof. Dr. Eckhard Spohr
Thermal and electrochemical oxidation reactions of alcohols on water-covered transition metal surfaces (TMOs) such as Co3O4 and CoFe2O4 spinels can nowadays be described best theoretically by a combination of quantum mechanical density functional theory (DFT), to study stable adsorbate species and transition states, and ab initio molecular dynamics (AIMD) based on DFT, to include ensemble effects and the impact of finite temperature. Ab initio MD studies in this project during the first funding period indicated that the presence of water on TMOs does not only play a significant role in determining surface structure (e.g., by lifting reconstructions), but also produces a large surface structure-dependent number of hydroxyl groups which are instrumental as hydrogen acceptors during electrochemical oxidation of 2-propanol to acetone. Furthermore, the mechanistic details of simulated oxidation reactions vary between those isolated 2-propanol molecules and larger adsorbed clusters of 2-propanol molecules (adsorbed and hydrated), and between different oxidation agents. We performed in-depth studies of the oxidation of 2-propanol to acetone on a hydrogen-deficient hydroxyl group-rich Co3O4 surface covered by a water phase, which may be characteristic for electrochemical oxidation reactions. Contrasting the observed behavior with specific molecular oxidants such as molecular oxygen or adsorbed OOH groups, or even the possibility that oxygen evolution competes with the oxidation of alcohols as evidenced by experimental investigations in projects A01 and A02, opens the exciting perspective to use our theoretical toolbox (in particular by extending DFT-based studies to the ones developed in project B08) to study the selectivity of alcohol oxidation reactions during the second funding period.
Based on the gained experience with 2-propanol oxidation, we plan to extend our scope to the study of different possible reaction channels of this molecule, and continue studying the influence of surface composition, surface defects, and hetero-atom doping, as and when more experimental data become available by a combination of DFT for a screening of mechanistic pathways evaluated by a modified volcano approach, and DFT-based AIMD for solvation and environmental effects. Additionally, we will widen our scope to study ethylene glycol, primarily because of its richer oxidation chemistry, but also because of the industrial importance of its oxidation products.
The long-term focus of this project is on the fundamental mechanistic aspects of alcohol oxidation on TMO surfaces, addressing how the electron-proton transfer steps can be manipulated by suitable environments with the goal to steer selectivity towards desired products.
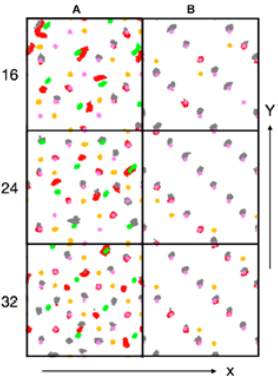
(Figure: Trajectory traces of surface atoms over 20 ps: (Co2+: green, Co3+: purple) and oxygen atoms of hydroxyl groups (red: oxygen originally from water molecule, orange: oxygen original from lattice) and water oxygen atoms (grey), taken from Front. Energy Res., 2020, 604799).

Copyright © TRR247 2026
Last update: Jan 25, 2026