![]()
![]()
![]()
![]()
Prof. Dr. Jörg Behler
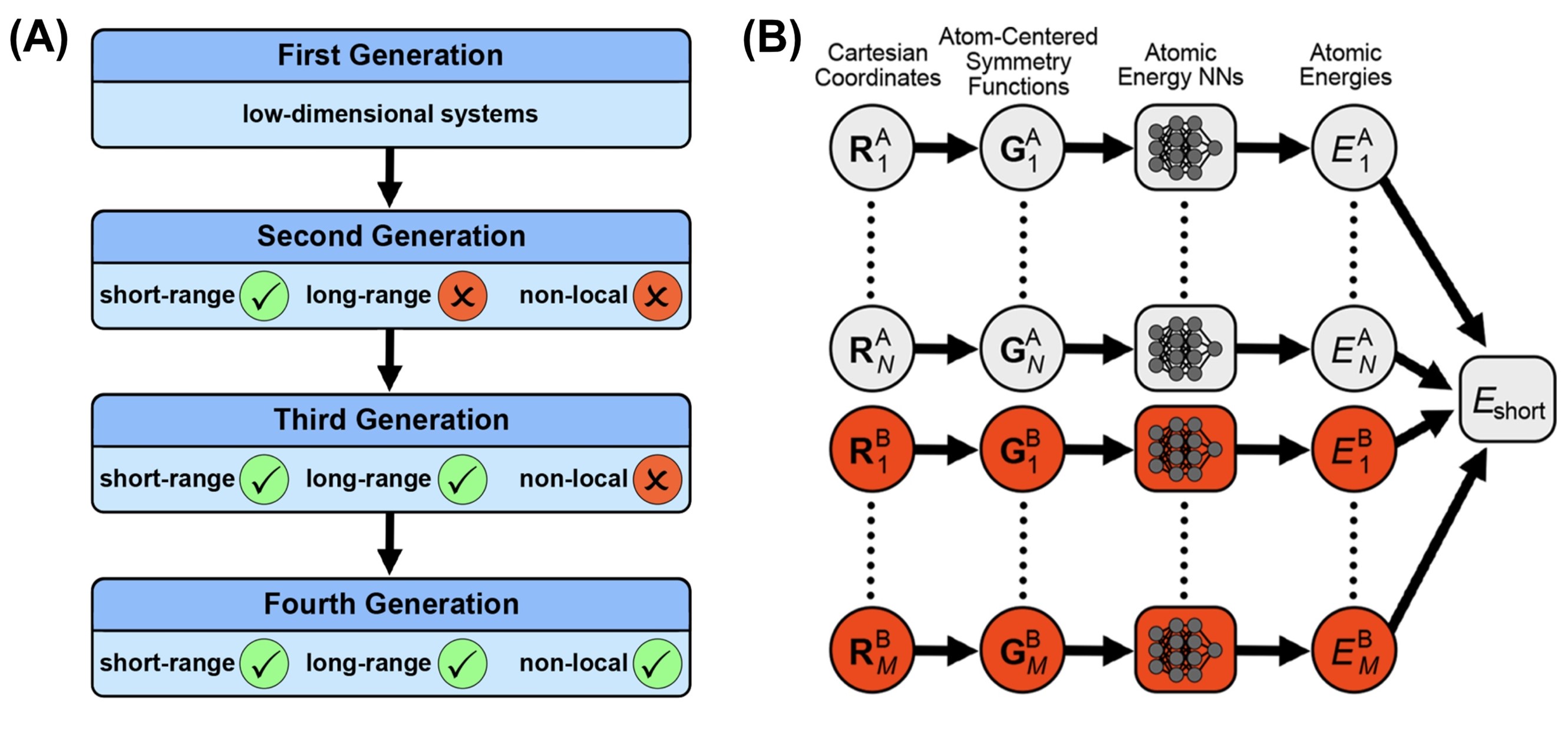
Nowadays, computer simulations have become an increasingly important tool, since in principle they allow to gain insights into the atomistic details of catalytic reactions. However, the reliability of the results obtained in these simulations crucially depends on the quality of the underlying potential-energy surface describing the atomic interactions. While the accuracy of first-principles calculations like density functional theory (DFT) is required for reliable and predictive simulations, the time and length scales of DFT-based ab initio molecular dynamics simulations are restricted by the high computational costs, which pose severe limitations on the complexity of the systems that can be studied.
To overcome this limitation, in recent years a lot of effort has been put in the development of machine learning potentials, which allow to combine the accuracy of electronic structure calculations with the efficiency of empirical potentials making them an important new tool in chemistry and materials science. Specifically, in the present project high-dimensional neural network potentials (HDNNPs), which we have developed and applied to many systems in the past one and a half decades, will be used to study the thermal oxidation of small alcohols in aqueous solution at Co3O4 surfaces, because this material has been identified as a very promising catalyst in the first funding period of the CRC.
Our aim is to first address the structural and dynamical properties of a variety of interfaces between Co3O4 and water including a wide range of facets, terminations, defects and surface morphologies as well as the possible formation of surface hydroxides. For this purpose, we will use molecular dynamics (MD) and metadynamics simulations based on HDNNPs trained to DFT+U data, which will allow to significantly extend the time and length scales to nanoseconds and tens of thousands of atoms. Once important structural interface features have been identified, their role as possible active sites will be assessed by studying their interaction and reactivity with 2-propanol in aqueous solution under different conditions like temperature and concentration in close collaboration with other theoretical and experimental groups in the CRC.
In the long-term perspective, in the third funding period the composition of the catalyst might be extended to doped cobalt spinel oxides, to gain insight into the relation between global as well as local composition and catalytic activity. Moreover, our established simulation setup will be applied to the oxidation of further alcohols like ethylene glycol to cover a broader range of systems included in the Comparative Study of the CRC.

Copyright © TRR247 2025
Last update: Aug 12, 2025